3 Derive novel groups
The utility augmentClusters.pl is used to derive novel sub-groups/sublineages within an existing classification of SARS-CoV-2 lineages/variants. The aim is to extend a target classification system by the incorporation of local/regional high genomic variants, which are used to infer/derive local variants of the virus. Users can specify the minimum size (minimum number of isolates included in the group) required for a novel group to be formed (–size), the minimum distance (in terms of number of characteristic high frequency genomic variants, –dist) between newly formed and extant groups, and a designations file, with the list of genomic variant characteristic of lineages/designations already included in the nomenclature (–deffile). The input consists in a metadata table in HaploCoV format and a list of genomic variant of hig frequency (genomic variant file). The output will a new designations file which will incorporate additional, novel designations/candidate lineages. The file will include all the extant lineages/variants specified in the metadata table, and also novel variants/lineages formed by the tool. All novel variants/lineages will be indicated by a suffix (–suffix) that can be specified by the user.
High frequencies alleles for Nexstrain data
Collections of high frequency alleles available from the HaploCoV Github repository are derived from the periodic processing of the complete collection of SARs-CoV-2 genomes included in the GISAID database; and hence should provide a more comprehensive representation of high frequency alleles than that which could be obtained by processing publicly available data re-distributed by Nexstrain with computeAF.pl. In the light of these considerations, users that have access only to Nextstrain data are kindly encouraged to take advantage (and use) high frequency allele files that are available from the HaploCoV repository instead of using computeAF.pl on their data. Please see above for how to download the most recent version of any of those files.
Options
augmentClusters.pl accepts the following parameters:
–metafile name of the metadata file (please see HaploCoV format for metdata ;
–posFile list of high frequency alleles (this is one of the main outputs of computeAF.pl, typically areas_list.txt);
–dist minimum edit distance (number of characteristic high frequency alleles) required for forming a novel group. Defaults to 2;
–suffix suffix used to delineate novel lineages. Defaults to N;
–size minimum size for a new subgroup within a lineage/group. Defaults to 100;
–tmpdir directory used to store temporary files;
–deffile file with lineage defining genomic variants. If linDefMut is specified, the most recent version will be downloaded;
--update update linDefmut to the most recent version? T=true. F=false. Default=T;
–oufile name of the output file;
The main output will be saved in the current folder.
Execution A command line for augmentClusters.pl should look something like:
perl augmentClusters.pl --outfile lvar.txt --metafile linearDataSorted.txt --posFile areas_list.txt
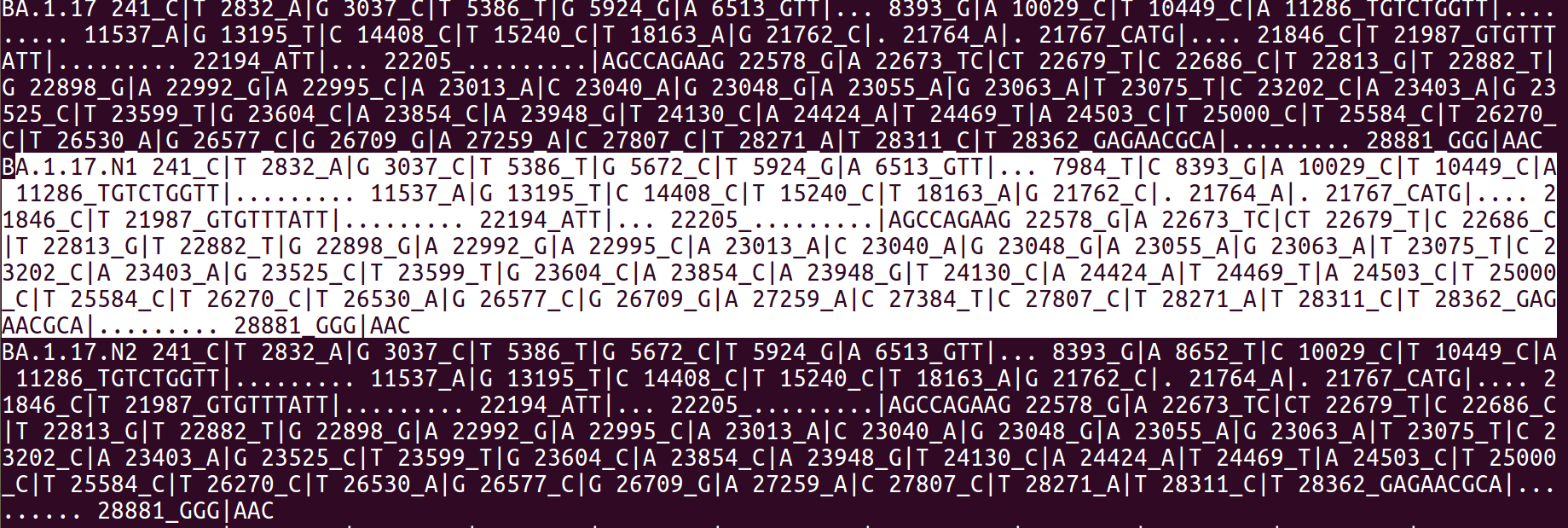
The main output file, lvar.txt will contain all current groups/lineages along with newly formed groups/sub-lineages, each designation will be reported in a distinct line, followed by the list of defining mutations. In HaploCoV we refer to this file format as designations file. An example is outlined in the screenshot below.
Warning
In augmentClusters.pl the –deffile parameter is used to provide a designations file. If no designation file is provided, characteristic/defining genomic variants are derived dynamically by processing the input metadata file (–metafile). This behaviour was implemented such as to avoid failures in the execution, however it might have some downsides: identifications/reconstruction of the genomic variants characteristic of a lineage will be based only on the data provided in input, and might result inconsistent across different executions. For these reasons we strongly advise users to provide a designations file with the –deffile option.
Warning
If/when linDefMut, the designations file of Pango lineages included in the HaploCoV repository is used, the –update option can be set to specify whether the most recent copy of the file should be downloaded (default, T=true), or wheter to use the copy already available in the current installation.
Please see below for a brief recap on **designations files* and their meaning.
Designations files
HaploCoV uses designations files to specify/list genomic variants that are characteristic of a group or lineage. The format of designations files is as follows: every line reports a lineage/group, defined by the corresponding id/name, followed by the list of characteristic genomic variants (defined here as those present in >50% of the isolates assigned to the group). Values are separated by spaces (see above). augmentClusters.pl provides its main output in designations files format, newly formed lineages/groups/sub-lineages in the output file are identified by a user specified suffix that a progressive number. The default value for this suffix is the letter “N”. If for example two novel lineages/groups/sub-lineages are derived in the Pango BA.1.17 lineage, these will be reported as:
in the output file (see above).