Things HaploCoV can do
The main aim of the tool is to facilitate the identification of novel variants/lineages of SARS-CoV-2 showing:
an increase in their prevalence (regional, national or global);
features associated with VOCs/VOIs (variants of concern or variants of interest);
both.
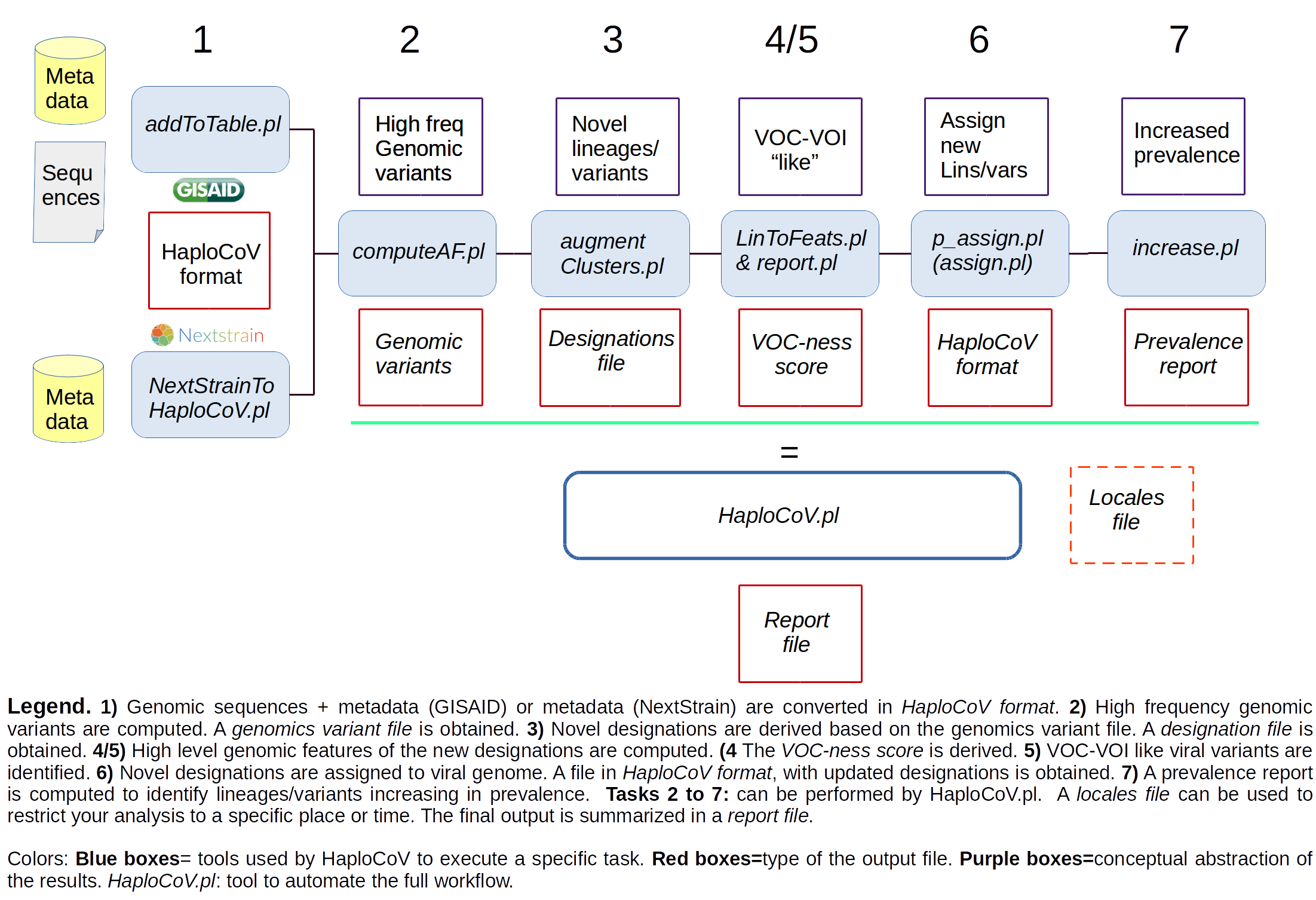
The tool incorporates a standalone scoring system (HaploCoV-score or VOC-ness score from here onward) for the identification and flagging of VOC and VOI-like variants based on the functional annotation of the genome. Interesting/relevant candidate variants are identified as those showing a significant increase (above a minimum threshold) in their score compared with their parental lineage/variant. The minimum threshold for significance was derived empirically (see the HaploCoV paper for more details).
Increase/decrease in prevalence is inferred by analyses of the (available) metadata. By default novel candidate variants with a prevalence above 1% in a region/country, and showing an increase by at least 2 fold over 4 weeks are reported. These parameters can be set by the user at runtime.
The main output consists in a report file that summarizes the prevalence and features (as defined by the criteria outlined above) of novel candidate variants of SARS-CoV-2.
The recommended way to execute HaploCoV is through HaploCoV.pl. This tool automates the execution of the complete workflow. However, users are also free to execute each single distinct task by themselves if they prefer. Each tool has its own entry in the manual.
The figure below illustrates the complete workflow. A complete description of each tool is found in the manual.