Where do I get the data
HaploCoV requires 3 input files:
the reference assembly of the SARS-CoV-2 genome in fasta format;
a multifasta file with SARS-CoV-2 genomes to be compared with the reference;
a .tsv file with metadata for the genome sequences in the multifasta.
Reference genome
HaploCoV can download the reference assembly of the SARS-CoV-2 genome for you when/if required (see the addToTable.pl utility for more details, here).
However, in the (hopefully) unlikely event that this should not work you can obtain a copy of the reference genome from:
on a unix system you can download this file, by:
wget https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/009/858/895/GCF_009858895.2_ASM985889v3/GCF_009858895.2_ASM985889v3_genomic.fna.gz
followed by:
gunzip GCF_009858895.2_ASM985889v3_genomic.fna.gz
Thie file (GCF_009858895.2_ASM985889v3_genomic.fna) is required by several tools in HaploCoV and needs to be in the same folder where our software package was installed (i.e. HaploCoV by default), if this file is missing tools will complain and halt their execution.
Genomes and metadata
GISAID
To run HaploCoV you need SARS-CoV-2 genome sequences and associated metadata.
Right now the GISAID EpiCoV database is the most complete and up to date point of access to obtain SARS-CoV-2 genomic data and associated metadata. Authorized users can download the complete collection of SARS-CoV-2 genome assemblies and their metadata by following the procedure illustrated in the figure below.
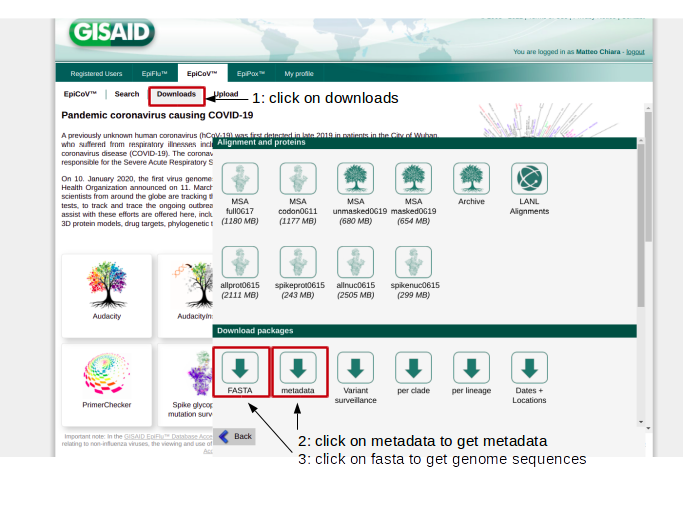
After de-compresson, 2 files should be obtained:
metadata.tsv a metadata table in .tsv format and;
sequences.fasta a multi-fasta file with SARS-CoV-2 genome sequences.
These files provide the main input to addToTable.pl; the utility in HaploCoV that extracts/formats all the data for subsequent analyses.
GISAID-Augur
Users that do not have full to the download of a complete dump of the GISAID EpiCoV database, can use the web interface to download a set of genomes of interest by selecting the corresponding entries. HaploCoV is compatible with the input format used by the Augur pipeline, hence users must download their selection of entries in the “Input for the Augur pipeline” format. See figure below.

Data will be downloaded in the form of a .tar archive. After unpacking two files should be obtained:
a .fasta file with the sequences: XXX.sequences.fasta;
and a .tsv file with metadata XXX.metadata.tsv.
These files can be provided in input to addToTable.pl.
Required metadata
Please be aware that some metadata are mandatory to execute HaploCoV and that column names in your metadata file MUST abide by the structure/names described below.
Mandatory metadata:
a valid unique identifier for every isolate, column name: “Virus name”;
a collection date, column name: “Collection date”;
a submission date, column name: “Submission date”;
location: the geographic place from where the isolated was collected, column name: “Location”;
a valid lineage/group/class associated with the genome. Column name: “Pango lineage” or “Lineage”.
Dates must be provided in YYYY-MM-DD format. Locations in the following format: Continent/Country/Region. Missing information must be indicated by NA (not available). Sequence identifiers can not be replaced by NA values.
An example of a valid metadata table is reported below.
Locales File Location
qualifier
start-date
end-date
genomic-variants
Italy
2022-11-01
2022-11-11
Europe/ Italy /Lombardy
BF.1
Thailand
2022-05-11
2022-11-26
Asia / Thailand / Buriram
BA.5
If your data were obtained from the GISAID EpiCoV database and following to the instructions provided above, all the requirements should be satisfied. Please notice that however, if any piece of data/columns (names indicated above must be matched exactly) is not found in your metadata table, the execution of addToTable.pl will halt and an error message will be raised.
Nextstrain
Users that do not have access to GISAID EpiCoV can obtain the complete collection of publicly available SARS-CoV-2 sequences and associated metadata from Nexstrain, please refer to here for more information. Metadata in “Nexstrain format” can be obtained from here. Since these data have already been processed by Nexstrain using their ncov workflow, genomic variants are already included in the metadata file and hence you will not need to download genomic sequences and align them to the reference genome. However, Nextstrain data still needs to be converted in HaploCoV format. This can be done by using NextStrainToHaploCoV.pl (see below).
Important: providing “custom” data
While HaploCoV was designed to work mainly with data from GISAID EpiCoV, the tool can in principle work also with data from other sources, including “custom” data that are not included in any repository. Please notice that however metadata must always comply with the format described above. Valid metadata tables must include 5 columns with the following names:
“Virus name”;
“Collection date”;
“Submission date”;
“Location”;
“Pango Lineage” or “Lineage”;
Names must match exactly, although mistakes in capitalisation are tolerated.